Коэффициенты уравнении ван дер ваальса. Уравнение Ван - дер - Ваальса. График уравнения Ван - дер - Ваальса. Примеры решения задач
Как мы уже упоминали, при низких температурах и высоких давлениях уравнение состояния идеального газа Менделеева – Клапейрона непригодно.
Учитывая собственный объём молекул и силы межмолекулярного взаимодействия, голландский физик И. Ван – дер – Ваальс (1837 – 1923 г.г.) вывел уравнение " реального газа ", используя две поправки для уравнения Менделеева – Клапейрона.
Учёт собственного объёма молекул. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объём других молекул, приводит к тому, что фактический свободный объём, в котором могут двигаться молекулы реального газа, будет равен не V μ (как в уравнении Менделеева – Клапейрона для одного моля газа), а V = (V μ -b) , где b – поправка на собственный объём молекул.
Можно показать, что поправка b равна учетверённому объёму молекул. Действительно, если, например, сближаются две молекулы, то центр любой из них не может приблизиться к центру другой молекулы на расстояние, меньшее диаметра d молекулы (оболочки молекул считаются непроницаемыми). Это означает, что для центров обеих молекул оказывается недоступным сферический объём радиуса d, т.е. объём, равный восьми объёмам молекулы или учетверённому объёму молекулы в расчёте на одну молекулу.
Учёт притяжения молекул. Поскольку при определённых расстояниях между молекулами действуют силы притяжения (а они, как мы уже говорили, проявляются раньше сил отталкивания), то их действие приводит к появлению " дополнительного " действия на молекулы " идеального " газа. Это давление Ван – дер – Ваальс назвал " внутренним " давлением. По модели "реального" газа вычисления показали, что " внутреннее " давление молекул обратно пропорционально квадрату молярного объёма, т.е.:
, (17.6)
где а – вторая постоянная (поправка) Ван – дер – Ваальса, характеризующая действие сил межмолекулярного притяжения, V μ – молярный объём газа.
Вводя эти поправки, получим итоговое уравнение Ван – дер – Ваальса для одного моля газа :
. (17.7)
Для произвольного количества вещества в ν молей газа (т.к. ν = m/M μ ) с учётом того, что V = ν V μ , уравнение Ван – дер – Ваальса примет вид:
 , (17.8)
, (17.8)
где поправки a и b – постоянные для каждого индивидуального газа величины, вычисляемые из экспериментальных данных (в простейшем случае записываются уравнения Ван – дер – Ваальса для двух известных из опыта состояний газа и решаются относительно величин a и b ).
Поскольку при выводе уравнения для " реального " газа Ван – дер – Ваальсом был сделан ряд весьма существенных упрощений, поэтому оно так же, как и уравнение Менделеева – Клапейрона является достаточно приближённым уравнением, которое, однако, лучше (особенно для не очень сильно сжатых газов) согласуется с опытом, чем уравнение состояния идеального газа.
Для более точного описания опытных данных для реальных газов пользуются эмпирическими уравнениями состояния, чаще всего уравнением Камерлинг – Оннеса, имеющим вид:
 , (17.9)
, (17.9)
которое построено с таким расчётом, чтобы всегда имелась возможность привести это уравнение к согласию с данными опыта простым вписыванием дополнительных членов без изменения формы уравнения. Коэффициенты B,C, F называются вириальными коэффициентами и представляются в виде многочленов, расположенных по возрастающим степеням Т -1 :
 , (17.10)
, (17.10)
и аналогично для коэффициентов C,D,E,F .
Как уже указывалось в § 60, для реальных газов необходимо учитывать размеры молекул и их взаимодействие друг с другом, поэтому модель идеального газа и уравнение Клапейрона-Менделеева (42.4) pV m =RT (для моля газа), описывающее идеальный газ, для реальных газов непригодны.
Учитывая собственный объем молекул и сил межмолекулярного взаимодействия, голландский физик И. Ван-дер-Ваальса (1837-1923) вывел уравнения состояния реального газа. Ван-дер-Ваальсом в уравнение Клапейрона-Менделеева введены две поправки.
1. Учет собственного объема молекул. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объем других молекул, сводится к тому, что фактический свободный объем, в котором могут двигаться молекулы реального газа, будет не V m , a V m - b , где b - объем, занимаемый самими молекулами. Объем b равен учетверенному собственному объему молекул. Если, например, в сосуде находятся две молекулы, то центр любой из них не может приблизиться к центру другой молекулы на расстояние, меньшее диаметра d молекулы. Это означает, что для центров обеих молекул оказывается недоступным сферический объем радиуса d, т. е. объем, равный восьми объемам молекулы, а в расчете на одну молекулу - учетверенный объем молекулы.
2. Учет притяжения молекул. Действие сил притяжения газа приводит к появлению дополнительного давления на газ, называемого внутренним давлением. По вычислениям Ван-дер-Ваальса, внутреннее давление обратно пропорционально квадрату молярного объема, т. е.
p" = a/V 2 m , (61.1)
где а- постоянная Ван-дер-Ваальса, характеризующая силы межмолекулярного притяжения, V m - молярный объем.
Вводя эти поправки, получим уравнение Ван-дер-Ваальса для моля газа (уравнение состояния реальных газов):
(p+a/V 2 m )(V m -b)=RT. (61.2)
Для произвольного количества вещества v газа (v =т/М) с учетом того, что V = vV m , уравнение Ван-дер-Ваальса примет вид
где поправки а и b - постоянные для каждого газа величины, определяемые опытным путем (записываются уравнения Ван-дер-Ваальса для двух известных из опыта состояний газа и решаются относительно а и b ).
При выводе уравнения Ван-дер-Ваальса сделан целый ряд упрощений, поэтому оно также весьма приближенное, хотя и лучше (особенно для несильно сжатых газов) согласуется с опытом, чем уравнение состояния идеального газа.
Уравнение Ван-дер-Ваальса не единственное уравнение, описывающее реальные газы. Существуют и другие уравнения, некоторые из них даже точнее описывают реальные газы, но не рассматриваются из-за их сложности.
§ 62. Изотермы Ван-дер-Ваальса и их анализ
Для исследования поведения реального газа рассмотрим изотермы Ван-дер-Ваальса - кривые зависимости р от V m при заданных Т, определяемые уравнением Ван-дер-Ваальса (61.2) для моля газа. Эти кривые (рассматриваются для четырех различных температур; рис. 89) имеют довольно своеобразный характер. При высоких температурах (T>T к) изотерма реального газа отличается от изотермы идеального газа только некоторым искажением ее формы, оставаясь монотонно спадающей кривой. При некоторой температуре Т к на изотерме имеется лишь одна точка перегиба К . Эта изотерма называется критической, соответствующая ей температура T к - критической температурой. Критическая изотерма имеет лишь одну точку перегиба К, называемую критической точкой; в этой точке касательная к ней параллельна оси абсцисс. Соответствующие этой точке объем V к и давление р к называются также критическими. Состояние с критическими параметрами (р к, V к , Т к ) называется критическим состоянием. При низких температурах (Т<Т к ) изотермы имеют волнообразный участок, сначала монотонно опускаясь вниз, затем монотонно поднимаясь вверх и снова монотонно опускаясь.


Для пояснения характера изотерм преобразуем уравнение Ван-дер-Ваальса (61.2) к виду
pV 3 m -(RT+pb) V 2 m +aV m -ab=0.
Уравнение (62.1) при заданных р и Т является уравнением третьей степени относительно V m ; следовательно, оно может иметь либо три вещественных корня, либо один вещественный и два мнимых, причем физический смысл имеют лишь вещественные положительные корни. Поэтому первому случаю соответствуют изотермы при низких температурах (три значения объема газа V 1 , V 2 и V 3 отвечают (символ «т» для простоты опускаем) одному значению давления р 1 ), второму случаю- изотермы при высоких температурах.
Рассматривая различные участки изотермы при Т<Т к (рис.90), видим, что на участках 1 -3 и 5-7 при уменьшении объема V m давление р возрастает, что естественно. На участке 3-5 сжатие вещества приводит к уменьшению давления; практика же показывает, что такие состояния в природе не осуществляются. Наличие участка 3-5 означает, что при постепенном изменении объема вещество не может оставаться все время в виде однородной среды; в некоторый момент должно наступить скачкообразное изменение состояния и распад вещества на две фазы. Таким образом, истинная изотерма будет иметь вид ломаной линии 7-6-2-1. Часть 7-6 отвечает газообразному состоянию, а часть 2-1 - жидкому. В состояниях, соответствующих горизонталь-
ному участку изотермы 6-2, наблюдается равновесие жидкой и газообразной фаз вещества. Вещество в газообразном состоянии при температуре ниже критической называется паром, а пар, находящийся в равновесии со своей жидкостью, называется насыщенным.
Данные выводы, следующие из анализа уравнения Ван-дер-Ваальса, были подтверждены опытами ирландского ученого Т. Эндрюса (1813-1885), изучавшего изотермическое сжатие углекислого газа. Отличие экспериментальных (Эндрюс) и теоретических (Ван-дер-Ваальс) изотерм заключается в том, что превращению газа в жидкость в первом случае соответствуют горизонтальные участки, а во втором - волнообразные.
Для нахождения критических параметров подставим их значения в уравнение (62.1) и запишем
p к V 3 -(RT к +p к b)V 2 +aV-ab= 0
(символ «т» для простоты опускаем). Поскольку в критической точке все три корня совпадают и равны V к , уравнение приводится к виду
p к (V-V к ) 3 = 0,
p к V 3 -3p к V к V 2 +3p к V 2 к V-p к V к = 0.
Так как уравнения (62.2) и (62.3) тождественны, то в них должны быть равны и коэффициенты при неизвестных соответствующих степеней. Поэтому можно записать
ркV 3 к =ab, 3р к V 2 к =а, 3p к V к =RT к +p к b. Решая полученные уравнения, найдем: V к = 3b, р к = а/(27b 2), T к =8a/(27Rb}.
Если через крайние точки горизонтальных участков семейства изотерм провести линию, то получится колоколообразная кривая (рис. 91), ограничивающая область двухфазных состояний вещества. Эта кривая и критическая изотерма делят

диаграмму р, V m под изотермой на три области: под колоколообразной кривой располагается область двухфазных состояний (жидкость и насыщенный пар), слева от нее находится область жидкого состояния, а справа - область пара. Пар отличается от остальных газообразных состояний тем, что при изотермическом сжатии претерпевает процесс сжижения. Газ же при температуре выше критической не может быть превращен в жидкость ни при каком давлении.
Сравнивая изотерму Ван-дер-Ваальса с изотермой Эндрюса (верхняя кривая на рис. 92), видим, что последняя имеет прямолинейный участок 2-6, соответствующий двухфазным состояниям вещества. Правда, при некоторых условиях могут быть реализованы состояния, изображаемые участками ван-дер-ваальсовой изотермы 5-6 и 2-3. Эти неустойчивые состояния называются метастабильными. Участок 2-3 изображает перегретую жидкость, 5-6 - пересыщенный пар. Обе фазы ограниченно устойчивы

При достаточно низких температурах изотерма пересекает ось V m , переходя в область отрицательных давлений (нижняя кривая на рис. 92). Вещество под отрицательным давлением находится в состоянии растяжения. При некоторых условиях такие состояния также реализуются. Участок 8 -9 на нижней изотерме соответствует перегретой жидкости, участок 9 - 10 - растянутой жидкости.
При высоких температурах последний член в (5) можно опустить, и тогда изотерма будет гиперболой, асимптотами которой являются изобара Р = 0 и изохора V = b .
Для исследования изотерм при любых значениях Т
умножим уравнение (4) на V
2 .
После раскрытия скобок уравнение изотермы примет вид ![]() (6)
(6)
Это уравнение третьей степени по V , в которое давление Р входит в качестве параметра. Поскольку его коэффициенты вещественны, уравнение имеет либо один вещественный корень, либо три корня. Каждому корню на плоскости (V,P ) соответствует точка, в которой изобара Р = const пересекает изотерму. В первом случае, когда корень один и точка пересечения будет одна. Так будет, как мы видели, при любых давлениях, если температура достаточно высока. Изотерма имеет вид монотонно опускающейся кривой MN .
 При более низких температурах и надлежащих значениях давления Р
уравнение (6) имеет три корня V
1 , V
2 , V
3 . В таких случаях изобара P = const
пересекает изотерму в трех точках L, C, G
(рис. 1). Изотерма содержит волнообразный участок LBCAG.
Она сначала монотонно опускается вниз (участок DB
), затем на участке BA
монотонно поднимается вверх, а за точкой A
снова монотонно опускается. При некоторой промежуточной температуре три корня V
1 , V
2 , V
3 становятся равными. Такая температура и соответствующая ей изотерма называются критическими
. Критическая изотерма FKH
всюду монотонно опускается вниз, за исключением одной точки K,
являющейся точкой перегиба изотермы. В ней касательная к изотерме горизонтальна. Точка K
называется критической точкой.
Соответствующие ей давление P k
, объем V k
и температура T k
называются также критическими.
Говорят, что вещество находится в критическом состоянии
, если его объем и давление (а следовательно, и температура) равны критическим.
При более низких температурах и надлежащих значениях давления Р
уравнение (6) имеет три корня V
1 , V
2 , V
3 . В таких случаях изобара P = const
пересекает изотерму в трех точках L, C, G
(рис. 1). Изотерма содержит волнообразный участок LBCAG.
Она сначала монотонно опускается вниз (участок DB
), затем на участке BA
монотонно поднимается вверх, а за точкой A
снова монотонно опускается. При некоторой промежуточной температуре три корня V
1 , V
2 , V
3 становятся равными. Такая температура и соответствующая ей изотерма называются критическими
. Критическая изотерма FKH
всюду монотонно опускается вниз, за исключением одной точки K,
являющейся точкой перегиба изотермы. В ней касательная к изотерме горизонтальна. Точка K
называется критической точкой.
Соответствующие ей давление P k
, объем V k
и температура T k
называются также критическими.
Говорят, что вещество находится в критическом состоянии
, если его объем и давление (а следовательно, и температура) равны критическим.
Для нахождения критических параметров P k , V k , T k учтем, что в критической точке уравнение (6) переходит в уравнение (7).
Поскольку в этом случае все три корня совпадают и равны V k , уравнение должно приводиться к виду (8).
Возводя в куб и сравнивая коэффициенты уравнений (7) и (8), получим три уравнения .
Решая их, найдем выражения для параметров критического состояния вещества: ![]() (9).
(9).
К тем же результатам можно прийти, заметив, что критическая точка К
является точкой перегиба изотермы, касательная в которой горизонтальна, а поэтому в точке К
должны соблюдаться соотношения  .
.
Решая эти уравнения совместно с уравнением изотермы (4) придем к формулам (9).
Не все состояния вещества, совместимые с уравнением Ван-дер-Ваальса, могут быть реализованы в действительности. Для этого необходимо еще, чтобы они были термодинамически устойчивы. Одно из необходимых условий термодинамической устойчивости физически однородного вещества состоит в выполнении неравенства . Физически оно означает, что при изотермическом увеличении давления объем тела должен уменьшаться. Иными словами, при возрастании V все изотермы должны монотонно опускаться. Между тем, ниже критической температуры на изотермах Ван-дер-Ваальса имеются поднимающиеся участки типа BCA (рис. 1). Точки, лежащие на таких участках, соответствуют неустойчивым состояниям вещества, которые практически реализованы быть не могут. При переходе к практическим изотермам эти участки должны быть выброшены.
Таким образом, реальная изотерма распадается на две ветви EGA и BLD , отделенные друг от друга. Естественно предположить, что этим двум ветвям соответствуют различные агрегатные состояния вещества. Ветвь EA характеризуется относительно большими значениями объема или малыми значениями плотности, она соответствует газообразному состоянию вещества. Напротив, ветвь BD характеризуется относительно малыми объемами, а следовательно, большими плотностями, она соответствует жидкому состоянию вещества . Мы распространяем, следовательно, уравнение Ван-дер-Ваальса и на область жидкого состояния. Таким путем удается получить удовлетворительное качественное описание явления перехода газа в жидкость и обратно.
Возьмем достаточно разреженный газ при температуре ниже критической. Исходное состояние его на диаграмме PV изображается точкой E (рис. 1). Будем сжимать газ квазистатически, поддерживая температуру T постоянной. Тогда точка, изображающая состояние газа, будет перемещаться по изотерме вверх. Можно было думать, что она достигает крайнего положения A , где изотерма обрывается. В действительности, однако, начиная с некоторой точки G , давление в системе перестает повышаться, и она распадается на две физически однородные части, или фазы : газообразную и жидкую.
Процесс изотермического сжатия такой двухфазной системы изображается участком GL горизонтальной прямой. При этом во время сжатия плотности жидкости и газа остаются неизменными и равными их значениям в точках L и G соответственно. По мере сжатия количество вещества в газообразной фазе непрерывно уменьшается, а в жидкой фазе - увеличивается, пока не будет достигнута точка L, в которой все вещество перейдет в жидкое состояние.
Эндрюс систематически исследовал ход изотерм углекислоты (СО 2) при различных температурах и на основе этих исследований ввел понятие критической температуры. Углекислота им была выбрана сознательно, так как она обладает критической температурой (31 0 С), лишь незначительно превышающей комнатную, и сравнительно невысоким критическим давлением (72,9 атм). Оказалось, что при температуре выше 31 0 С изотермы углекислоты монотонно опускаются вниз, т.е. имеют гиперболический вид. Ниже этой температуры на изотермах углекислоты появляются горизонтальные участки, на которых изотермическое сжатие газа приводит к его конденсации, но не к увеличению давления. Таким путем было установлено, что сжатием газ можно превратить в жидкость только тогда, когда его температура ниже критической.
При специальных условиях могут быть реализованы состояния, изображаемые участками изотермы GA и BL. Эти состояния называются метастабильными. Участок GA изображает так называемый пересыщенный пар , участок BL - перегретую жидкость . Обе фазы обладают ограниченной устойчивостью. Каждая из них может существовать до тех пор, пока она не граничит с другой более устойчивой фазой. Например, пересыщенный пар переходит в насыщенный, если в него ввести капли жидкости. Перегретая жидкость закипает, если в нее попадают пузырьки воздуха или пара.
Уравнение Менделеева - Клапейрона является уравнением состояния идеального газа и довольно точно описывает поведение реальных газов при небольшой плотности, т.е. достаточно низком давлении и высокой температуре ( ![]() ).
).
При понижении температуры и увеличении давления, плотность газа увеличивается, а расстояние между его молекулами уменьшается, поэтому пренебрегать их объёмом и взаимо-
Рис. 23 действием мы не можем.
Силы взаимного притяжения между молекулами направлены внутрь газа, т. е. в сторону наибольшего окружения периферийных молекул (рис.23).
Действие этих сил подобно наличию некоторого добавочного давления на газ, называемого внутренним.
В связи с тем, что молекулы газа занимают конечные размеры, они занимают суммарный объём V / . Поэтому объём, предоставленный для передвижений молекулам, будет меньше на величину V" . Таким образом, для описания состояния реальных газов необходимо сделать две поправки:
а ) на дополнительное давление, обусловленное взаимодействием молекул;
б ) на уменьшение объёма, в связи с учётом размеров самих молекул.
Возьмём за основу уравнение состояния идеального газа и, внеся в него соответствующие поправки, получим уравнение состояния реального газа. Для одного моля газа имеем
Внесённые поправки были впервые рассчитаны и предложены Ван-дер-Ваальсом (гол.)
где а и в – постоянные Ван-дер-Ваальса.
Уравнение Ван-дер-Ваальса для одного моля реального газа имеет вид:
![]() . (26)
. (26)
![]() Учитывая, что и, умножив обе части уравнения на , получим уравнение Ван-дер-Ваальса для любой массы газа:
Учитывая, что и, умножив обе части уравнения на , получим уравнение Ван-дер-Ваальса для любой массы газа: ![]() . (27)
. (27)
Полученные нами уравнения имеют третью степень относительно V , например, для одного моля после преобразования, оно будет иметь вид:
0.
Это означает, что оно может иметь либо три действительных, либо один действительный и два мнимых корня, при чём физический смысл имеют только действительные корни.

 Эти особенности уравнения состояния нашли своё отражение в графиках зависимости p
(V m
), называемых кривыми Ван-дер-Ваальса
(рис. 24).
Эти особенности уравнения состояния нашли своё отражение в графиках зависимости p
(V m
), называемых кривыми Ван-дер-Ваальса
(рис. 24).
Заметим, что при некоторой температуре лишь одна точка перегиба. Она называется критической .
Предпринималось много попыток для учета отклонений свойств реальных газов от свойств идеального газа путем введения различных поправок в уравнение состояния идеального газа. Наибольшее распространение вследствие простоты и физической наглядности получило уравнение Ван-дер-Ваальса (1873).
Первая поправка в уравнении состояния идеального газа рассматривает собственный объем, занимаемый молекулами реального газа. В уравнении Дюпре (1864)
p (V – nb ) = nRT (1.3)
постоянная b учитывает собственный мольный объем молекул.
При понижении температуры межмолекулярное взаимодействие в реальных газах приводит к конденсации (образование жидкости). Межмолекулярное притяжение эквивалентно существованию в газе некоторого внутреннего давления (иногда его называют статическим давлением). Изначально величина была учтена в общей форме в уравнении Гирна (1865)
(p + ) (V – nb ) = nRT . (1.4)
Ван-дер-Ваальс в 1873 г. дал функциональную интерпретацию внутреннего давления. Согласно модели Ван-дер-Ваальса, силы притяжения между молекулами (силы Ван-дер-Ваальса ) обратно пропорциональны шестой степени расстояния между ними, или второй степени объема, занимаемого газом. Считается также, что силы притяжения суммируются с внешним давлением. С учетом этих соображений уравнение состояния идеального газа преобразуется в уравнение Ван-дер-Ваальса:
 (1.5)
(1.5)
или для одного моля
 . (1.6)
. (1.6)
Значения постоянных Ван-дер-Ваальса a и b , которые зависят от природы газа, но не зависят от температуры, приведены в таблице 1.3.
Таблица 1.3. Постоянные Ван-дер-Ваальса для различных газов
| Газ | a , л 2 *бар* моль -2 | b,см 3 * моль -1 | Газ | a, л 2 * бар* моль -2 | b, см 3 * моль -1 |
| He | 0,03457 | 23,70 | NO | 1,358 | 27,89 |
| Ne | 0,2135 | 17,09 | NO 2 | 5,354 | 44,24 |
| Ar | 1,363 | 32,19 | H 2 O | 5,536 | 30,49 |
| Kr | 2,349 | 39,78 | H 2 S | 4,490 | 42,87 |
| Xe | 4,250 | 51,05 | NH 3 | 4,225 | 37,07 |
| H 2 | 0,2476 | 26,61 | SO 2 | 6,803 | 56,36 |
| N 2 | 1,408 | 39,13 | CH 4 | 2,283 | 42,78 |
| O 2 | 1,378 | 31,83 | C 2 H 4 | 4,530 | 5,714 |
| Cl 2 | 6,579 | 56,22 | C 2 H 6 | 5,562 | 63,80 |
| CO | 1,505 | 39,85 | C 3 H 8 | 8,779 | 84,45 |
| CO 2 | 3,640 | 42,67 | C 6 H 6 | 18,24 | 115,4 |
Уравнение (1.6) можно переписать так, чтобы выразить в явном виде давление
![]() (1.7)
(1.7)
или объем
![]() (1.8)
(1.8)
Уравнение (1.8) содержит объем в третьей степени и, следовательно, имеет или три действительных корня, или один действительный и два мнимых. При высоких температурах уравнение (1.8) имеет один действительный корень, и по мере повышения температуры кривые, вычисленные по уравнению Ван-дер-Ваальса, приближаются к гиперболам, соответствующим уравнению состояния идеального газа.
На рис. 1.4 (стр. 7) приведены изотермы, вычисленные по уравнению Ван-дер-Ваальса для диоксида углерода (значения констант a и b взяты из табл. 1.3). Из рисунка видно, что при температурах ниже критической (31,04 °С) вместо горизонтальных прямых, соответствующих равновесию жидкости и пара, получаются волнообразные кривые 12345 с тремя действительными корнями, из которых только два, 1 и 5, физически осуществимы. Третий корень (точка 3) физически не реален, поскольку находится на участке кривой 234, противоречащем условию стабильности термодинамической системы . Состояния на участках 12 и 54, которые соответствуют переохлажденному пару и перегретой жидкости, соответственно, являются неустойчивыми (метастабильными) и могут быть лишь частично реализуемы в специальных условиях. Так, осторожно сжимая пар выше точки 1 (рис. 1.4), можно подняться по кривой 12. Для этого необходимо отсутствие в паре центров конденсации, и в первую очередь пыли. В этом случае пар оказывается в пересыщенном, т.е. переохлажденном состоянии. И наоборот, образованию капелек жидкости в таком паре способствуют, например, попадающие в него ионы. Это свойство пересыщенного пара используется в известной камере Вильсона (1912), применяемой для регистрации заряженных частиц. Движущаяся заряженная частица, попадая в камеру, содержащую пересыщенный пар, и соударяясь с молекулами, образует на своем пути ионы, создающие туманный след – трек, который фиксируется фотографически.
Согласно правилу Максвелла (the Maxwell construction ), которое имеет теоретическое обоснование, для того, чтобы расчетная кривая соответствовала экспериментальной равновесной изотерме, нужно вместо кривой 12345 провести горизонтальную прямую 15 так, чтобы площади 1231 и 3453 были равны. Тогда ордината прямой 15 будет равна давлению насыщенного пара, а абсциссы точек 1 и 5 – мольным объемам пара и жидкости при данной температуре.
По мере повышения температуры все три корня сближаются, и при критической температуре T c все три корня становятся равными. В критической точке изотерма Ван-дер-Ваальса имеет точку перегиба с горизонтальной касательной , то есть
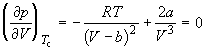 , (1.9)
, (1.9)
 . (1.10)
. (1.10)
Совместное решение этих уравнений дает:
что позволяет определять константы уравнения Ван-дер-Ваальса из критических параметров газа. Соответственно, согласно уравнению Ван-дер-Ваальса, критический фактор сжимаемости Z c для всех газов должен быть равен
![]() (1.14)
(1.14)
Из таблицы 1.2 видно, что хотя значение Z c для реальных газов приблизительно постоянно (0,27 – 0,30 для неполярных молекул), оно все же заметно меньше вытекающего из уравнения Ван-дер-Ваальса. Для полярных молекул наблюдается еще большее расхождение.
Принципиальное значение уравнения Ван-дер-Ваальса определяется следующими обстоятельствами:
1) уравнение было получено из модельных представлений о свойствах реальных газов и жидкостей, а не явилось результатом эмпирического подбора функции f(p,V,T), описывающей свойства реальных газов;
2) уравнение долго рассматривалось как некоторый общий вид уравнения состояния реальных газов, на основе которого было построено много других уравнений состояния (см. ниже);
3) с помощью уравнения Ван-дер-Ваальса впервые удалось описать явление перехода газа в жидкость и проанализировать критические явления. В этом отношении уравнение Ван-дер-Ваальса имеет преимущество даже перед более точными уравнениями в вириальной форме (см. 1.1, 1.2).
Причиной недостаточной точности уравнения Ван-дер-Ваальс считал ассоциацию молекул в газовой фазе, которую не удается описать, учитывая зависимость параметров a и b от объема и температуры, без использования дополнительных постоянных. После 1873 г. сам Ван-дер-Ваальс предложил еще шесть вариантов своего уравнения, последнее из которых относится к 1911 г. и содержит пять эмпирических постоянных. Две модификации уравнения (1.5) предложил Клаузиус, и обе они связаны с усложнением вида постоянной b . Больцман получил три уравнения этого типа, изменяя выражения для постоянной a . Всего известно более сотни подобных уравнений, отличающихся числом эмпирических постоянных, степенью точности и областью применимости. Выяснилось, что ни одно из уравнений состояния, содержащих менее 5 индивидуальных постоянных, не оказалось достаточно точным для описания реальных газов в широком диапазоне p, V, T, и все эти уравнения оказались непригодными в области конденсации газов. Из простых уравнений с двумя индивидуальными параметрами неплохие результаты дают уравнения Дитеричи и Бертло (см. табл. 1.4).